Subclustering with PopPIPE¶
Overview¶
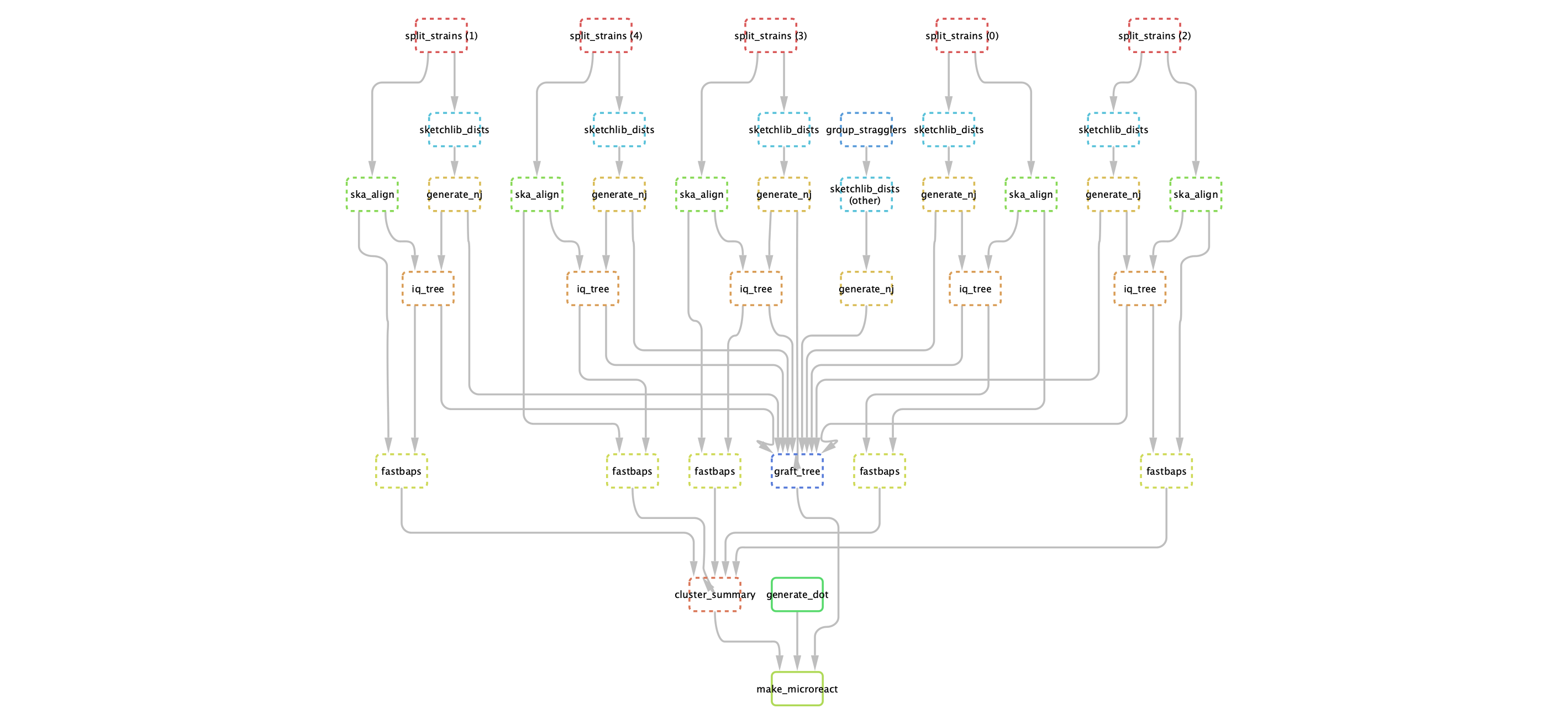
You can run PopPIPE on your PopPUNK output, which will run subclustering and visualisation within your strains. The pipeline consists of the following steps:
Split files into their strains.
Calculate core and accessory distances within each strain.
Use the core distances to make a neighbour-joining tree.
(lineage_clust mode) Generate clusters from core distances with lineage clustering in PopPUNK.
Use ska to generate within-strain alignments.
Use IQ-TREE to generate an ML phylogeny for each strain using this alignment, and the NJ tree as a starting point.
Use fastbaps to generate subclusters which are partitions of the phylogeny.
Create an overall visualisation with both core and accessory distances, as in PopPUNK. The final tree consists of refining the NJ tree by grafting the maximum likelihood trees for subclusters to their matching nodes.
An example DAG for the steps (excluding ska index, for which there is one per sample):
Installation¶
PopPIPE is a snakemake pipeline, which depends upon snakemake and pandas:
conda install snakemake pandas
Other dependencies will be automatically installed by conda the first time you run the pipeline. You can also install them yourself and omit the -use-conda directive to snakemake:
conda env create -n poppipe --file=environment.yml
Then clone the repository:
git clone git@github.com:johnlees/PopPIPE.git
Usage¶
Modify
config.ymlas appropriate.Run
snakemake --cores <n_cores> --use-conda.
On a cluster or the cloud, you can use snakemake’s built-in --cluster argument:
snakemake --cluster qsub -j 16 --use-conda
See the snakemake docs for more information on your cluster/cloud provider.
Alternative runs¶
For quick and dirty clustering and phylogenies using core distances from pp-sketchlib alone, run:
snakemake --cores <n_cores> --use-conda lineage_clust
To create a visualisation on microreact:
snakemake –use-conda make_microreact
Config file¶
PopPIPE configuration¶
script_location: Thescripts/directory, if not running from the root of this repositorypoppunk_db: The PopPUNK HDF5 database file, without the.h5suffix.poppunk_clusters: The PopPUNK cluster CSV file, usuallypoppunk_db/poppunk_db_clusters.csv.poppunk_rfile: The--rfileused with PopPUNK, which lists sample names and files, one per line, tab separated.min_cluster_size: The minimum size of a cluster to run the analysis on (recommended at least 6).
IQ-TREE configuration¶
enabled: Set tofalseto turn off ML tree generation, and use the NJ tree throughout.mode: Set tofullto run with the specified model, set tofastto run using--fast(like fasttree).model: A string for the-mparameter describing the model. Adding+ASCis recommended.
fastbaps configuration¶
levels: Number of levels of recursive subclustering.script: Location of therun_fastbapsscript. Find by runningsystem.file("run_fastbaps", package = "fastbaps")in R.
Updating a run¶
Running snakemake from the same directory will keep outputs where possible,
so new additions will automatically be included.
TODO: How to do this when adding new isolates with poppunk_assign